
Tags | |
UUID | 19a44aae-f145-11e9-8682-bc764e2038f2 |
Fractional Distillation
From ChemPRIME
When a pure substance is mixed with another pure substance in which it is soluble, the substances become completely interspersed at the molecular level. In thinking about making solutions at the molecular level an analogy to a can of marbles may be useful. In the analogy, a layer of red marbles is placed in the bottom of a can and covered with a second layer of white marbles. After shaking the can for a short time, the marbles are mixed randomly.
Now let us imagine that you want to collect all the red marbles again. If you simply shake the can, it is unlikely that you will ever divide the marbles into two layers, each with only one kind of marble. Similarly, if two miscible liquids are combined, a chemist cannot simply unmix the liquids into pure components.
Continuing the analogy, what if a few green marbles and blue marbles are placed into the can? Given enough red and white marbles, it may be difficult to determine that the green marbles and blue marbles are actually there. Similarly, when chemists have a multi-component solution which may contain traces of important chemical species, they are faced with the challenge of detecting whether these chemicals are present in solution.
To deal with these difficulties, chemists employ different methods to separate solutions into their components. One of these essential techniques is called distillation.
It is immediately apparent from the cartoon in the section on Raoult’s law that the composition of the vapor in equilibrium with a solution and the composition of the solution itself must be different. While only 50 percent of the molecules in the liquid phase are benzene molecules, in the vapor phase they represent 80 percent of the molecules. This difference in composition between the two phases is the reason we can separate components of a liquid mixture by distillation.
In the laboratory, distillation is usually carried out in an apparatus similar to that shown in figure 1. The liquid mixture to be distilled is heated in a round-bottom flask until it boils. The emerging vapor travels into the condenser where it is cooled sufficiently to return to the liquid state. The condensed liquid can then be collected in a suitable container. In practice this container is usually changed several times during the course of the distillation. Each sample collected in this way is called a fraction, and the whole process is called fractional distillation. A thermometer may be used to determine when vapor reaches the condenser, and to determine the progression of the distillation.
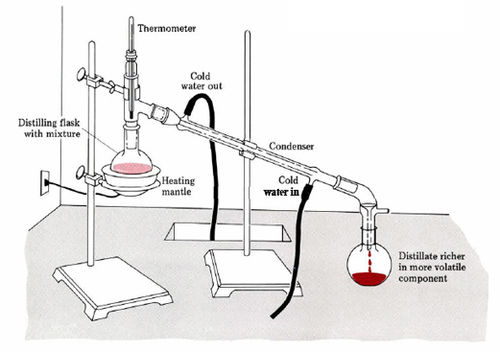
Figure 12. Schematic of a distillation apparatus. The mixture is heated and begins to boil. Cold water run through the condenser tube, causing the gaseous mixture to return to the liquid phase. A more pure volatile component is collected in a flask at the other end of the condenser tube
Suppose we use the distillation apparatus shown in the figure to distill an equimolar mixture of benzene and toluene. We would first have to raise the temperature of the mixture to 92.4°C, at which point the total vapor pressure would be 1 atm (101.3 kPa) and the liquid would boil. At 92.4°C the vapor in equilibrium with an equimolar mixture has a mole fraction of benzene equal to 0.71 (slightly different from that in the cartoon on the page discussing Raoult’s law). Accordingly, when our mixture is distilled, vapor of this composition will be condensed and the first drops in the collection vessel will contain 71 percent benzene molecules and 29 percent toluene molecules. We will now have a sample which is much richer in benzene than the original. In theory we could go on repeating this process indefinitely. If we redistilled a liquid mixture having χb =0.71, it would boil at 87°C and yield a condensed vapor whose mole fraction of benzene would be 0.87. A third distillation would yield benzene with χb =0.95, and a fourth would yield a product with χb =0.99. If we went on long enough, we could obtain a sample of benzene of any desired purity.
In practice repeated distillation takes up a lot of time, and we would lose part of the sample each time. Fortunately there is a way out of this difficulty. We insert a glass column filled with round beads, or with specially designed packing, between the distillation flask and the condenser. When a vapor reaches the bottom-most glass beads en route to the condenser, part of it will condense. This condensate will be richer in the less volatile component, toluene, and so the vapor which passes on will be richer in benzene than before. As it passes through the column, the vapor will undergo this process several times. In effect the fractionating column allows us to perform several distillations in one operation. A well-designed laboratory column makes it possible to effect several hundred distillations in one pass. By using such a column, it would be possible to distill virtually pure benzene from the equimolar mixture, leaving virtually pure toluene behind.
Fractional distillation is used on a very large scale in the refining of petroleum. Some of the large towers seen in petroleum refineries are gigantic fractionating columns. These columns enable the oil company to separate the petroleum into five or six fractions called cuts. The more-volatile cuts can be used directly for gasoline, kerosene, etc. The less-volatile cuts contain the very long-chain alkanes and are usually converted into shorter-chain alkanes by cracking, as described in the section on unsaturated hydrocarbons.
While fractional distillation can be used to separate benzene and toluene and most mixtures of alkanes which show only small deviations from ideal behavior, it cannot be used to separate mixtures which exhibit a maximum or a minimum in the vapor-pressure versus mole-fraction curve. Thus any attempt to separate benzene and methanol (seen in the graph on the page discussing Raoult’s law) by fractional distillation does not result in methanol distilling over initially but rather results in a mixture with χCH3OH =0.614. A mixture of this composition is not only more volatile than methanol or benzene but also has a higher vapor pressure than any other mixture, as can be seen from that graph. This is why it distills first. Such a mixture is called a minimum boiling azeotrope. Conversely, for a mixture such as chloroform and acetone which exhibits large enough negative deviations from Raoult’s law, a maximum boiling azeotrope of low volatility distills at the end of a fractional distillation, preventing complete separation of the pure components.
This Collection is empty
- Comments
- Attachments
- Stats
No comments |